Berlin (Labnews Media LLC) – Ein neues maschinelles Lernverfahren namens Euclidean Fast Attention (EFA) kann globale Atomwechselwirkungen in chemischen Systemen deutlich effizienter darstellen. Das könnten künftige Simulationen in der Chemie und Materialwissenschaft genauer und schneller machen und die Entwicklung neuer Medikamente, effizienterer Batterien sowie nachhaltiger Materialien beschleunigen.
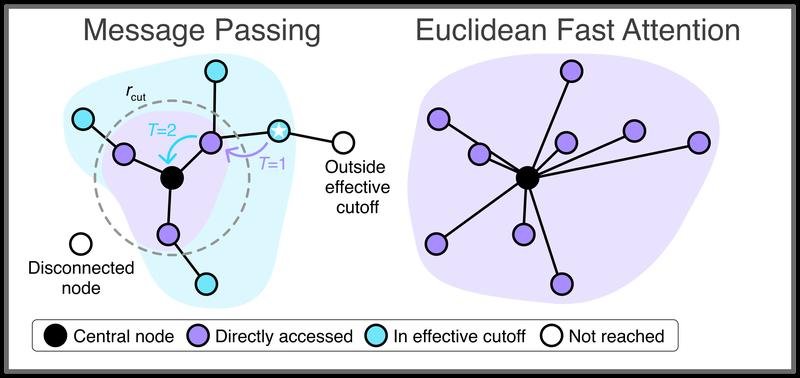
Forschende von Google DeepMind Berlin, dem Berlin Institute for the Foundations of Learning and Data (BIFOLD) und der Technischen Universität Berlin haben das Verfahren entwickelt. Es löst ein zentrales Problem bisheriger Methoden: Die herkömmliche Self-Attention skaliert quadratisch mit der Anzahl der Atome und wird bei größeren Molekülen schnell zu rechenintensiv. EFA skaliert dagegen linear, ist speziell für den euklidischen Raum optimiert und wahrt wichtige physikalische Symmetrien.
Die Methode erfasst langreichweitige Effekte zuverlässig, bei denen konventionelle maschinelle Lern-Kraftfelder häufig Fehler machen. Damit lassen sich komplexe Vielteilchensysteme mit Hunderten oder Tausenden Atomen realistischer simulieren. Prof. Klaus-Robert Müller vom BIFOLD und der TU Berlin hob hervor, dass das Verfahren einen wichtigen Schritt zu quantenmechanisch präziseren Modellen vielteiliger Systeme darstelle.
Die Studie mit dem Titel „Machine Learning Global Atomic Representations with Euclidean Fast Attention“ wurde im März 2026 in der Fachzeitschrift Nature Machine Intelligence veröffentlicht. Sie gilt als vielversprechender Ansatz, um maschinelle Lernmethoden für anspruchsvolle chemische und materialwissenschaftliche Simulationen robuster und effizienter zu machen.